
This is an explanation of false positives in qRT-PCR: what they are and how they occur. qRT-PCR is the type of PCR used to test for COVID-19 and it stands for ‘Semi-Quantitative Reverse Transcriptase-Polymerase Chain Reaction’ (a bit of a mouthful, so sometimes people just say ‘The PCR test’). To unpack qRT-PCR and understand its use(s), we need to step back and think about genetic information and PCR in the round.
Your genes and how PCR amplifies tiny amounts of DNA
The genetic information of many species is made of DNA. This is true (as far as we know) for all bacteria, fungi, protozoa, plants, insects and vertebrates, including you. If you need to study the DNA from a small sample of one of these, you can amplify part of it using PCR (polymerase chain reaction – we’ll come to the ‘RT’ part in a minute). This is shown in Figure 1a. PCR is incredibly sensitive. It can start with as little as a single DNA molecule and quickly (in a couple of hours) amplify part of it to produce billions and billions of copies – enough to study in depth. PCR is used widely in research, clinically and in forensic medicine: genomic DNA in a tiny blood stain can be amplified by PCR so that investigators can combine it with other tests to show whether the blood came from Suspect A or Suspect B.
To tell (in general) if PCR has worked, the potential PCR products can be separated and visualised on a gel system. The gel tells you how long the PCR product is, which gives you an idea of whether you’ve amplified the intended target (which doesn’t always happen), and it contains a negative control to show that the PCR hasn’t amplified an unintended product, such as a contaminant. Both are very important and are considered below.
Today’s PCR is often measured by machines rather than gel systems. With a combination of light, detectors and clever fluorescent dye chemistry, it is possible for the machine to detect PCR amplification as it occurs, in ‘real time’. Real time PCR is sometimes referred to as qPCR, for semi-quantitative PCR. qPCR is immensely powerful, because it can tell you not only whether any amplifiable DNA was present in your starting material, but if all goes well, how much – it’s quantitative. However, it does not tell you what the amplified PCR product was; machines can be blind to the nature of the product. Without careful calibration, this can be a problem.
PCR only works on DNA
You may have noticed that the genetic information list above excluded viruses. Although the genomes of many viruses – like ‘flu – are also made of DNA, the genetic information of others is different: they use a related molecule called RNA. Coronaviruses (eg COVID-19) are in this second group: they use RNA as their genetic material. But PCR doesn’t work on RNA – it requires DNA. So the PCR of Figure 1a won’t work to amplify COVID-19 genetic material, which is made of RNA.
Fortunately, there is a work-around for this: we can first make a DNA copy of the RNA. This brings us to the ‘RT’ in ‘RT-PCR’. In RT-PCR, there is an initial step in which the enzyme reverse transcriptase (RT) uses RNA to make a DNA copy, and then PCR can use this copy as shown in Figure 1a. In certain circumstances, RT-PCR can be evaluated in a ‘real time’ way (qRT-PCR) using machines, as described above. This is a broadly adopted and powerful approach in research. It can tell molecular biologists how much of a certain type of RNA is present in a sample, for example whether a human biopsy contains an unhealthy level of cancer-associated RNA. A similar qRT-PCR approach is also taken to determine whether samples contain the COVID-19 virus genome. Given that much is at stake with this approach, it is probably wise to be aware of the challenges to, and limitations of qRT-PCR in general.
qRT-PCR as a double-edged sword
The amazing sensitivity of methods based on PCR is both their exoneration and their potential downfall. Each PCR cycle doubles the amount of material, which may not sound impressive, but it really is. To illustrate this, imagine you were perched on top of the Big Ben tower (96 metres up) and it doubled in length every second. Within 22 seconds (22 doublings), you would be travelling at the speed of light (leaving aside Special Relativity). So if something goes wrong in the PCR, you quickly amplify an aberrant result to staggering proportions. Although it varies, researchers are typically interested in PCR amplification that becomes detectable within 25-35 cycles (doublings), referred to as the threshold value (Ct) and discussed below. Let’s now turn to some of the issues in qRT-PCR that research laboratories are punctiliously careful to ensure – or guard against.
Specificity of the PCR primers
Primer specificity is critical, because you wish to amplify your sequence of interest, nothing else. Although represented here simplistically, this specificity has a lot to do with shape of the primer and it is, alas, not a binary thing. Figure 1b shows how primers can bind to regions of DNA that have a similar shape to the target, but are different. This binding is less efficient than binding to the bona fide, intended target but there are many more ‘target look-alikes’ than intended targets, so given enough time, primer binding to non-targets will occur. Primer binding is also affected by the chemical composition of the PCR reaction. In research laboratories, this can be carefully standardised, but it is more difficult to do so where samples come from different sources that are each unique, such as the nose or throat contents of people screened population-wide. Note that undesired primer binding events only have to occur twice, because the resulting PCR product then has primer sequences at each end that make a perfect match for subsequent cycles. If the PCR reaction has products of different sizes as it goes along, the shorter one tends to predominate, so if the undesired primer binding generates a short product, it will be amplified preferentially – but the PCR machine won’t tell you this. PCR reactions containing multiple primer pairs (‘multiplexing’) have a greater chance of producing undesired products because there are even more ‘target look-alikes’.
Reverse transcriptase: the ‘RT’ in qRT-PCR
Reverse transcriptase (RT) is a sensitive enzyme and goes ‘off’ easily, so it must be stored at a low temperature. Good research labs validate it in every experiment. But on the other hand, supposing the reverse transcriptase is working when there is little RNA present in the sample but a lot of DNA. Is the reverse transcriptase attracted to make a copy of DNA, perhaps rather than RNA? Yes, it is. So how much DNA do we each contain?
DNA and RNA in qRT-PCR samples
With some basic assumptions, the genetic material (DNA) in most human cell nuclei is 12.8 billion bases long. If you work out how much this represents in an entire person – you – it is truly amazing: there is enough DNA in each of us (more or less) to make 431 round trips to… the sun (from Droitwich). Even if 100 genomes-worth of COVID-19 virus RNA existed per cell and it was efficiently copied by reverse transcriptase, there would be over 4,000x more genomic DNA. Where possible, and in general, research laboratories therefore take careful steps to purify the RNA or remove DNA before beginning the qRT-PCR protocol. This doesn’t always work ideally, so there are important checks, such as running the reaction without the RT step; if you detect a product in this situation, you know something’s gone wrong, because PCR doesn’t work on RNA – the RT step should be critical. An additional complication is that healthy human cells contain as much as 5x more RNA than DNA. All of this means that human samples contain ample material for off-target amplification.
Cycle number
Most research applications limit the number of qRT-PCR cycles. More technically speaking, this is determined by the threshold cycle number – or Ct – which is the PCR cycle number at which there is enough product to give a signal that is above the background level. In research, Ct values are often well under 30 and those much over 35 may merely reflect background levels. (It’s slightly more complicated because a PCR signal is more likely to be detected the more starting material there is, so research scientists determine the relative amount of product; we’ll skip this here, as it doesn’t affect false positives.) Increasing the cycle number also increases the chance of detecting non-specific primer binding. Critically, PCR machines do not distinguish between ‘false’ signals and those that come from intended targets during PCR; the machine measures product levels, not what the product is. Only with a gel system, sequencing or some other method, does the nature of the PCR product become clearer and these checks are used in research, particularly when setting up an experiment. Otherwise, the machine can happily register a product that has nothing to do with your intended target, and you will never know without additional checks.
Contamination with amplifiable DNA
This is an extraordinary facet of the sensitivity of PCR and it plagues all laboratories that use it, especially ones that often amplify the same type of PCR product from different samples. The nature of PCR contamination can seem magical to beginners: somehow, a vanishingly tiny amount of product from last week’s PCR got into today’s. And this contamination can be caused by as little as a single molecule. Contamination is detected by negative controls, such as a PCR reaction where you do not expect any product, like setting everything up but leaving out the sample or the reverse transcriptase. But because contamination is such a major pain, by far the best approach is to avoid contamination in the first place, rather than detecting it after it happened. For this reason, special precautions are taken in most laboratories to avoid contamination, such as using aerosol barrier tips (which are expensive) for pipetting, frequently changing gloves, using dedicated materials and working in a regularly cleaned and dedicated area. This is expensive. If there is contamination, experiments have to stop until it is rooted out. This is time-consuming, frustrating, can take days and can also be costly. Solutions and enzymes have to be tested and discarded if they are suspected of being a source. Pipettes and machines can also be the source of contamination and have to be purged if this turns out to be the case. Contamination is sometimes blatant, such as when all of the PCR reactions give products although only some (or none) were expected to. But it can be subtle because the amount of contaminating material is lower; in these cases, only some PCR reactions produce a product due to contamination. These are difficult to detect and require trouble-shooting experience as they can, of course, critically alter the interpretation of the data.
Concluding comments
This is a technical summary stripped of as much jargon as possible. As it relates to COVID-19, it doesn’t cover so-called ‘cold’ positives, in which virus RNA (including RNA fragments) is present in samples that do not contain viable or infectious virus and yet may still may give a positive signal. But it should highlight that although qRT-PCR is immensely powerful in research, its potential pitfalls require punctilious safeguards. In research, each experiment is performed with independent samples on at least two occasions – a minimal requirement for publication by respected journals. Interpreting both positive and negative qRT-PCR results requires experience that is most abundant among molecular biologists working on eukaryotic systems, and one wonders to what extent they have been called upon to advise on COVID-19 testing. There are few technical grounds on which to be confident that qRT-PCR is readily scalable, but doubts about its clinical application could be met squarely, whilst respecting patient anonymity, by complete, contemporaneous and auditable transparency.
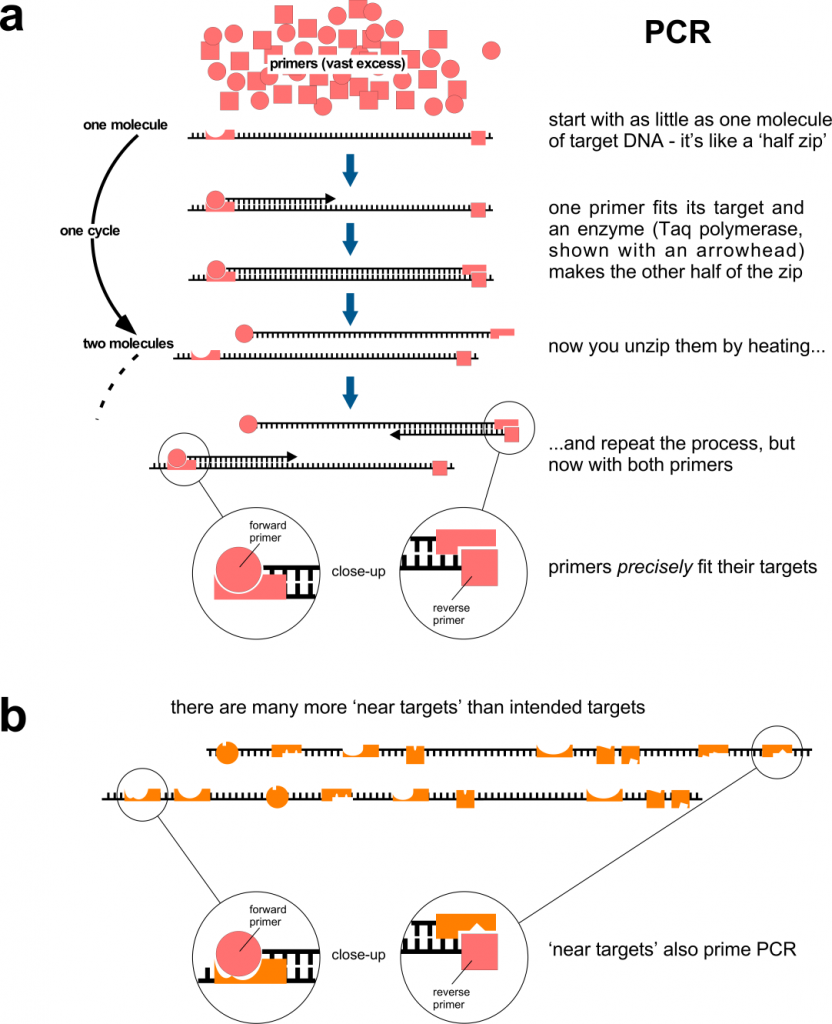
Figure 1. Polymerase chain reaction, PCR. a, The amount of PCR primers is unlimiting and the primers usually match their target perfectly. The PCR principle works in qRT-PCR. b, In an ideal world, each primer is specific for its intended target, but in reality they can match ‘target look-alikes’. Although these don’t work as well as the intended targets, there are many more look-alikes (than intended targets) and binding to them on only two occasions may be sufficient to produce an unintended (‘false’) positive.
The author is a research scientist working on eukaryotic molecular biology who has a PhD in microbial pathogenicity and has been using RT-PCR for over 30 years.










Donate
We depend on your donations to keep this site going. Please give what you can.
Donate TodayComment on this Article
You’ll need to set up an account to comment if you don’t already have one. We ask for a minimum donation of £5 if you'd like to make a comment or post in our Forums.
Sign UpLatest News
Next PostLatest News